5. Deep Dive · Systemic Inflammation & Fibrosis in RDEB
RDEB is, at its core, a disease of continuous inflammatory immune activation, leading to some of the most challenging parts of the disease (Not to be relied on for investment or patient care)

Co authored by Dr Alexander Nystrom (research group leader at the Department of Dermatology in Freiburg), Marouan Zarrouk from Debra Research, Nataliya Migulina & Ivan Fedyunin.
This article will focus on inflammation and fibrosis in RDEB. Read on for:
Science snapshot
Fibrosis – When Inflammation Becomes Permanent Damage
Key burdens of chronic inflammation in RDEB
Patient journey & health‑economics
Standard‑of‑care — and its limitations
Pipeline & emerging science (Rheacell, Mission EB, Losartan, JAKi, etc)
Opportunities & challenges
Market size
Investor take-aways
1. Science snapshot
Recessive Dystrophic Epidermolysis Bullosa (RDEB) is often described as a skin-fragility disorder, but that misses the bigger picture. The constant blistering and open wounds in the absence of collagen 7 (C7) don’t just damage the skin — they set off a relentless inflammatory cascade that affects the whole body. Every blister is an acute injury.
Keratinocytes and resident immune cells release pro-inflammatory cytokines, including IL-1⍺ and β (interleukin-1 alpha and beta), which trigger the recruitment and activation of additional immune cells through inflammatory signaling pathways. Moreover, C7 is more than a structural protein, its loss triggers dysregulation of pro-fibrotic and pro-inflammatory signals – of which heightened TGF-β (transforming growth factor beta ) activation in the dermal wound environment might be particularly important in the context of fibrosis. Studies show C7-deficient fibroblasts compared to normal fibroblast tend to display enhanced TGF-β activity including increased TGF-β cytokine expression, along with other altered cytokines, while addition of exogenous C7 can normalize their behavior (Nyström et al., 2013; Akasaka et al., 2021).
This cascade of events lights up across the wound bed resulting in a dermal microenvironment characterized by constant wound signaling: high levels of TGF-β, IL-1, IL-6, TNF (tumor necrosis factor) and other cytokines secreted or shedded by cells, potentially resulting in ongoing NF-κB and JAK/STAT signalling pathway activation, and an aberrant mix of fibroblast phenotypes. Persistently elevated levels of cytokines contribute to increased recruitment of neutrophils, macrophages, and pro-inflammatory phenotypic shifts of fibroblasts creating a positive feedback loop of chronic inflammation that fails to resolve. Further production of chemokines like IL-8/CXCL8 attracts waves of neutrophils, which in RDEB linger far beyond their welcome, releasing proteases and oxidants that cause more tissue damage. Elevated TGF-β drives fibroblasts to differentiate into myofibroblasts that alter the extracellular matrix protein composition and contract the tissue, leading to thick scarring. Thus, RDEB is not only a disease of skin fragility but also of dysregulated wound repair.
This isn’t just a local problem. Severe RDEB patients can have dozens of wounds open at once, each releasing a plethora of pro-inflammatory signals into circulation. One of the key drivers of systemic inflammation in RDEB is the cytokine IL-6 (interleukin-6), which as a potential biomarker of the disease correlates with total wound surface area and severity of the disease and acts as a potent amplifier of local injury signals from the wounded skin (Reimer-Taschenbrecker et al., 2024). Elevated IL-6 levels trigger the liver to ramp up production of acute-phase proteins such as CRP (C-reactive protein) and SAA (serum amyloid A). IL-6 may also skew T-cell responses toward a pro-inflammatory Th17, and enhances fibroblast activation and matrix remodelling, contributing to relentless fibrosis and contractures (Johnson et al., 2020). Crucially, IL-6 does not act alone – other inflammatory cytokines like IL-1s, TNF, and IL-17A are also elevated. Together they create a feedback loop that keeps the immune system in a state of perpetual alert. The result is a form of chronic systemic inflammation that contributes to anaemia, growth impairment, and metabolic decline — and in severe cases, complications like AA amyloidosis (a secondary amyloid disease) and cardiac failure (for example, due to inflammation-associated cardiomyopathy).
Adding fuel to the fire, the chronic open wounds in RDEB together with likely intrinsic inability to counteract bacterial challenges, result in changes of the skin’s normal microbiome, which further amplifies inflammation. Staphylococcus aureus quickly becomes dominant overgrowing normal flora. This may directly or indirectly promote activation of inflammasome pathways in resident skin cells and immune cells that leads to elevation in the release of IL-1β and other pro-inflammatory signals. In healthy skin, inflammation resolves. In RDEB, the triggers never stop — so neither does the inflammatory response.
RDEB is, at its core, a disease of continuous inflammatory immune activation. The skin injury is the spark, but it’s the unrelenting, dysregulated inflammation that drives much of the day-to-day burden, accelerates tissue decline, and shapes the course of the disease.
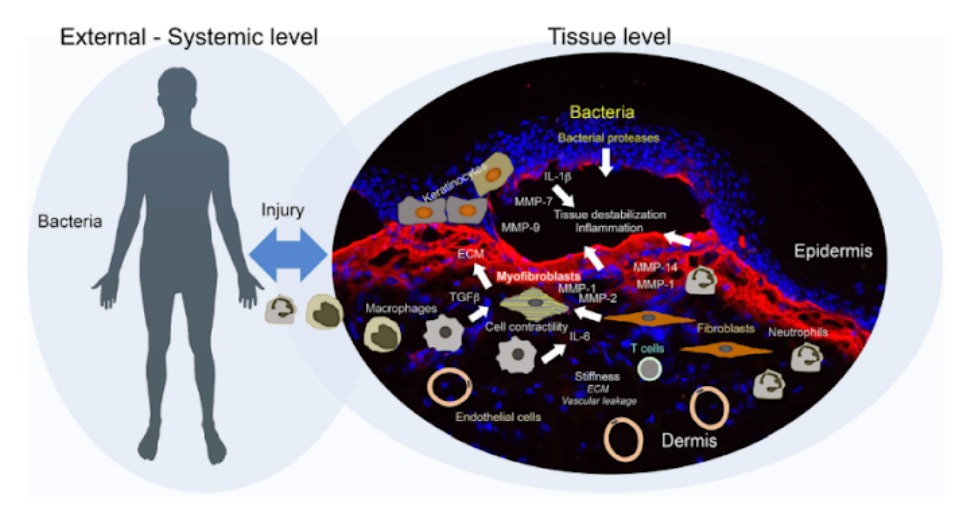
Fig.1. Secondary disease mechanisms and potential disease modifiers in DEB (Kiritsi et al., 2021).
2. Fibrosis – When Inflammation Becomes Permanent Damage
In RDEB, chronic inflammation doesn’t just damage tissue — it rewires the skin’s repair machinery.
In normal wound healing, fibroblasts migrate into the wound bed to deposit and subsequently mature the extracellular matrix (ECM), secrete growth factors that promote angiogenesis and keratinocyte proliferation to support re-epithelialization. Fibroblasts may transiently differentiate into contractile α-SMA⁺ (alpha-smooth muscle actin positive) myofibroblasts to help contract the wound. Once repair is complete, myofibroblasts undergo apoptosis, inflammation resolves, and the ECM is remodelled so that the healed skin gains near to normal elasticity.
In RDEB, loss of C7 and constant mechanical injury reprogram fibroblasts into pathological states. NF-κB–primed “inflammatory fibroblasts” secrete high levels of IL-6, IL-1β, and chemokines, perpetuating immune recruitment. At the same time, the wound contains ECM proteins including thrombospondin-1 which activates TGF-β driving fibroblasts into persistent highly contractile and profibrotic myofibroblasts, which in non-healing wounds and chronically injured tissue fail to undergo apoptosis and instead accumulate. Such fibroblasts are contractile and this together with the nature of the ECM they deposit lead to a stiffened tissue . The stiffened matrix stiffness further sustains TGF-β signaling, locking fibroblasts into a self-perpetuating fibrosis loop. The result is chronic, non-resolving wounds that not only impair skin function but also create a tumor-permissive stroma favoring squamous cell carcinoma.
Fibrosis in RDEB is not just a cosmetic issue. It drives life-altering deformities:
Mitten hands: Progressive scarring fuses fingers together, impairing fine motor function.
Joint contractures: Tightened, fibrotic skin restricts movement and can lock joints in painful positions.
Oesophageal strictures: Scar tissue in the oesophagus narrows the passage, making swallowing difficult and leading to malnutrition.
Over years, the inflammation–fibrosis cycle also creates a tumour-promoting microenvironment in people with RDEB. Activated fibroblasts behave much like carcinoma-associated fibroblasts in cancer, releasing growth factors, matrix-remodelling enzymes, and cytokines that encourage keratinocyte proliferation and mutation. This “primed” stroma is one reason cutaneous squamous cell carcinoma in RDEB is unusually aggressive & remains the leading cause of death in these patients.
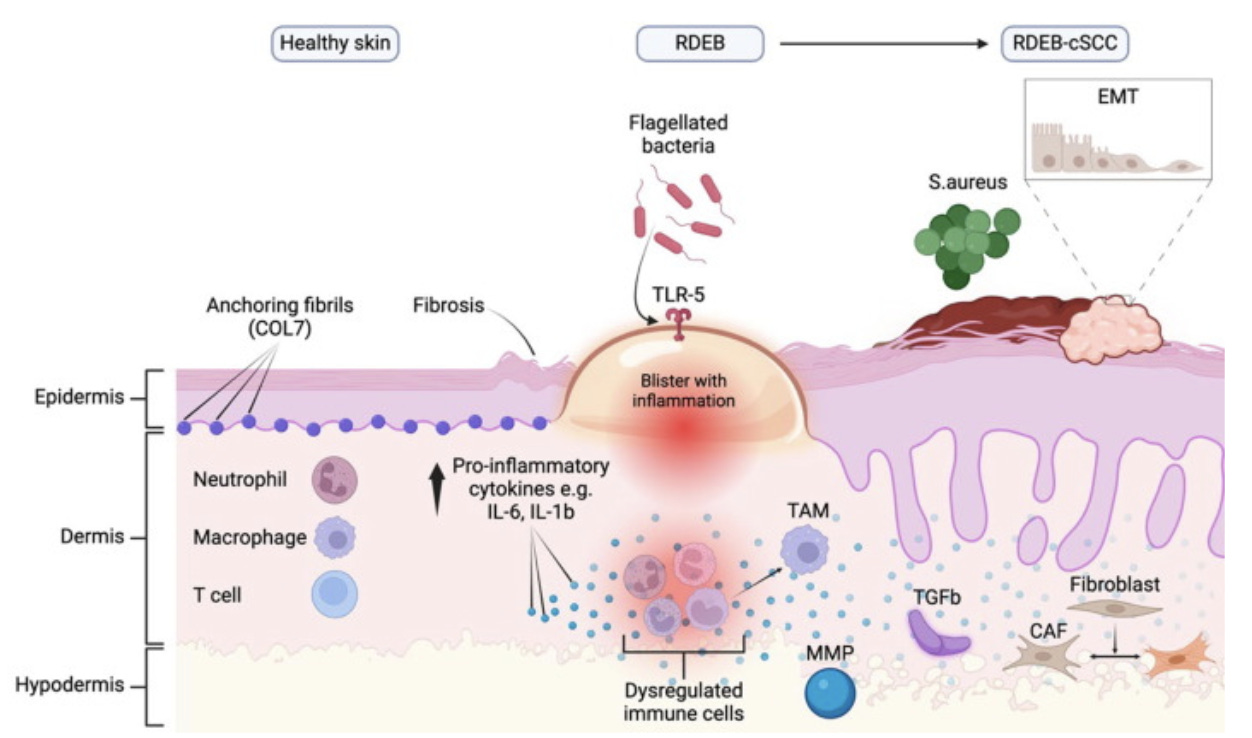
Fig. 2. Summary and comparisons of the microenvironment of healthy skin, RDEB skin and RDEB-cSCC skin. Abbreviation: CAF, cancer-associated fibroblast; COL7, collagen VII; EMT, epithelial to mesenchymal transition; IL, interleukin; MMP, matrix metalloproteinase(s); RDEB-cSCC, recessive, dystrophic epidermolysis bullosa associated cutaneous squamous cell carcinoma(s); RDEB, recessive dystrophic epidermolysis bullosa; S.aureus, staphylococcus aureus; TAM, tumour-associated macrophage(s); TGF-β, transforming growth factor beta; TLR, toll-like receptor(s) (Jacków-Malinowska et al., 2024)
3. Key burdens of chronic inflammation in RDEB
Beyond damaging the skin, inflammation in RDEB places a constant, measurable burden on the body. It’s not just a byproduct of blistering — it’s a driver of many downstream complications.
Key burdens of chronic inflammation in RDEB:
Delayed wound healing: Repeated injury with excessive inflammation drives inflammatory cell-influx and fibroblast activation, causing dysregulated ECM breakdown and deposition and fibrosis. This self-sustaining loop stalls healing and locks wounds in a chronic state.
Pain and itch: Cytokines and chemokines sensitise nerve endings, intensifying pain and causing relentless pruritus.
Nutritional and metabolic strain: Systemic inflammation increases energy expenditure, drives muscle breakdown, and suppresses appetite, worsening malnutrition.
Anaemia of chronic disease: IL-6–induced hepcidin release limits iron availability for red blood cell production.
Growth and hormonal impacts: Chronic inflammation disrupts endocrine signalling, slowing growth and delaying puberty.
Organ complications: Sustained high SAA can cause AA amyloidosis; cytokine-driven effects can impair cardiac and renal function.
Fibrosis and scarring: Inflammatory cytokines, particularly IL-1β and TGF-β1, activate fibroblasts and myofibroblasts, leading to progressive dermal fibrosis and contractures.
Infection susceptibility: Barrier loss and immune dysregulation increase the frequency and severity of secondary infections.
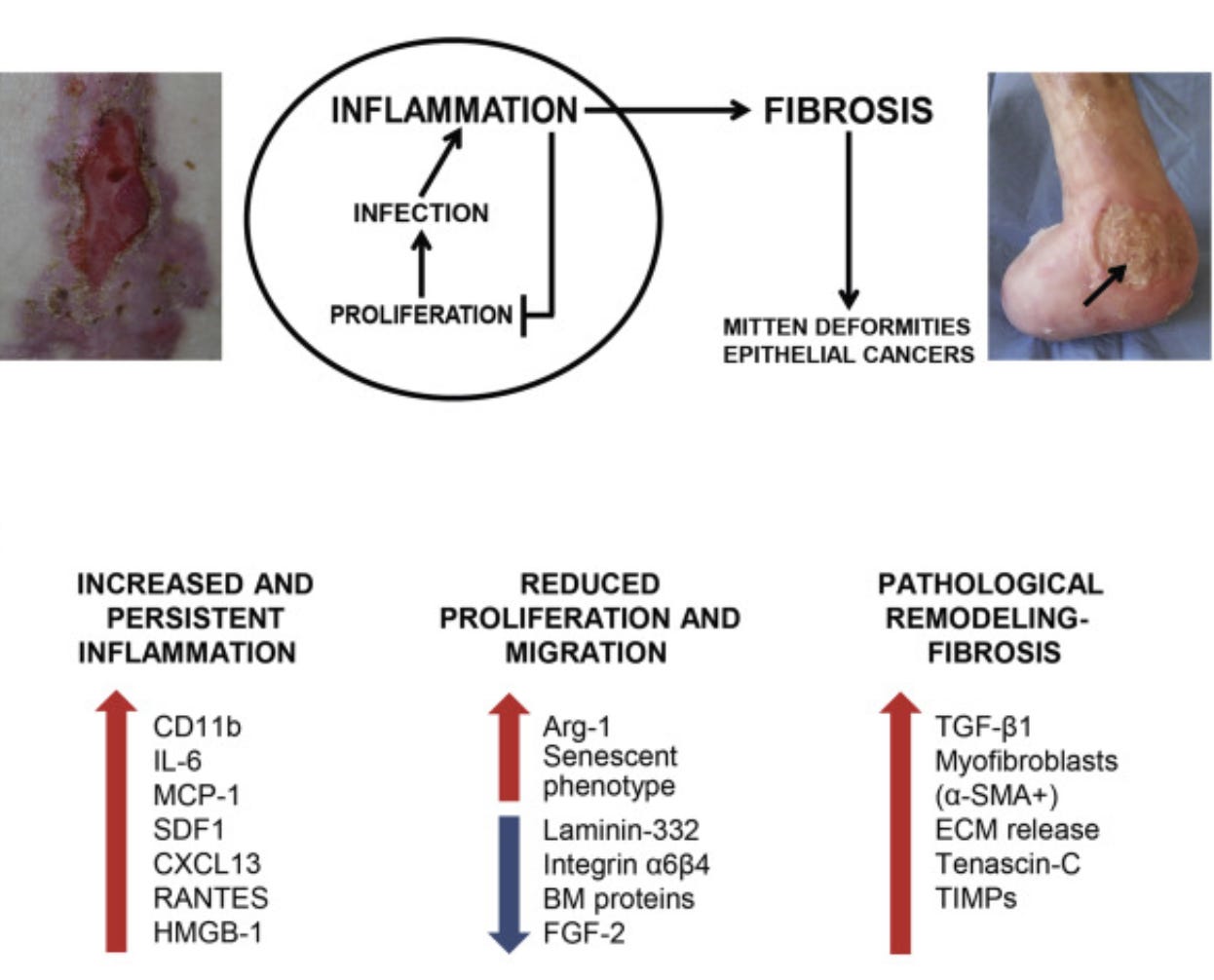
Fig. 3. Skin blistering and alterations affecting skin repair in dystrophic epidermolysis bullosa (DEB). The vicious cycle of skin repair in RDEB. An image of a nonhealing bleeding wound located over a bony prominence surrounded by erythematous inflamed skin is shown in the left panel. The image in the right panel shows mitten foot deformity with digit loss of a patient with severe RDEB. A deep chronic wound with raised edges and fibrinous exudate adhering to the wound bed is also visible on the malleolus (arrow). Bottom panel: molecular alterations found in RDEB skin/wounded skin and their effects in the different phases of the healing process. α-SMA, α-smooth muscle actin; FGF, fibroblast growth factor; MCP, monocyte chemoattractant protein; RANTES, regulated on activation normal T cell expressed and secreted; SDF, stromal cell–derived factor; TIMP, tissue inhibitor of metalloproteinase (Odorisio et al., 2017).
4. Patient journey & health‑economics
Inflammation and fibrosis together drive a care model that is both high-frequency and high-intensity. Costs accumulate from:
Chronic wound care: full-body dressing changes for up to 3 hours/daily with Specialized dressings are extremely costly (up to $250k annually depending on the severity); lifetime care runs into millions. Nursing time, antimicrobials and clinic visits add up.
Surgical interventions: Fibrotic deformities drive repeated procedures, including multiple hand releases (often with grafts and intensive aftercare, costing tens of thousands per surgery) and recurrent esophageal dilations.
Frequent hospitalisations: for infection, nutritional crisis, or acute complications. The average cost of a single inpatient stay in the US is over $14,000, with RDEB admissions are often longer.
Oncology care: By age 35–40, over 70% of severe RDEB patients develop SCC due to lifelong scar carcinogenesis, patients may undergo several surgeries and treatments each year, causing oncology-related expenses to accumulate rapidly.
Multidisciplinary care: dermatology, gastroenterology, surgery, ophthalmology, dentistry, nutrition, physiotherapy. Mean costs are shown in figure 4 while for severe cases they can climb dramatically.
Indirect costs: loss of employment, need for home adaptations, mobility aids, and caregiver burnout.
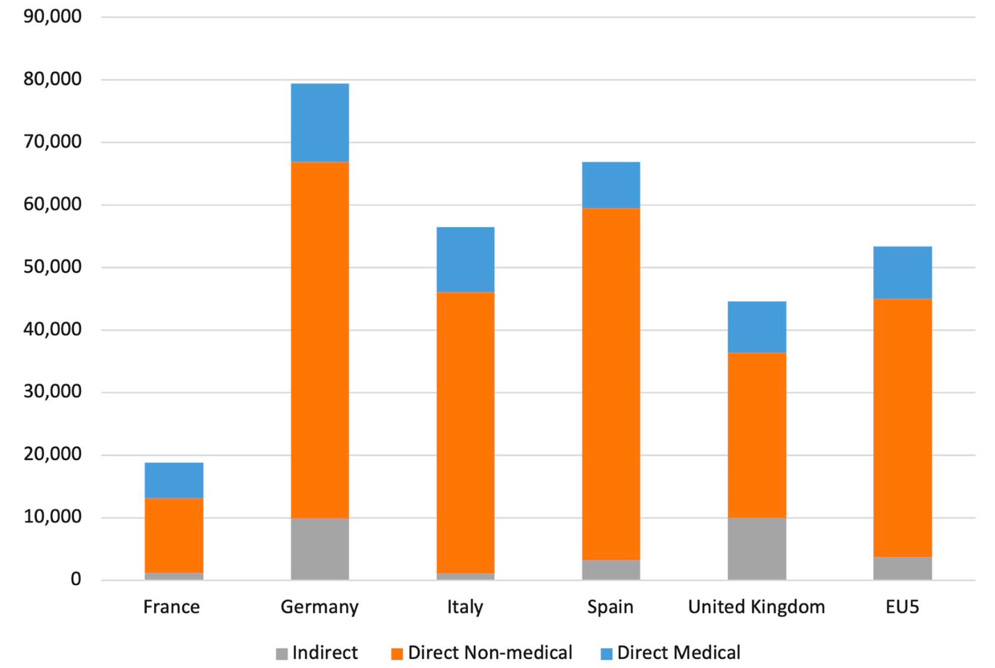
Fig. 4. Mean annual costs for all dystrophic epidermolysis bullosa patients across countries (2020, €). For severe patients the costs can be much higher. From Angelis et. al. Orphanet J Rare Dis, 2022, 17(1):346.
Because these processes are self-perpetuating, even partial suppression of inflammation or modulation of fibrotic signaling could delay high-cost complications and meaningfully reduce lifetime expenditure. This is a rare case where pathway-targeted therapy has the potential to change both survival curves and economic curves.
5. Standard‑of‑care — and its limitations
For inflammation and fibrosis in RDEB, the current standard-of-care is almost entirely supportive and symptomatic. There are no approved therapies that directly target the cytokine cascades or fibrotic pathways driving disease progression. Management today aims to reduce triggers, mitigate complications, and maintain function for as long as possible.
Current approaches
Wound care: Meticulous dressing regimens to reduce friction, prevent infection, and maintain a moist healing environment. While this can reduce acute inflammation from trauma, it does not modulate the underlying inflammatory signalling.
Infection control: Topical and systemic antibiotics to treat secondary infections, which can spike inflammatory burden. These address microbial triggers but not sterile inflammation.
Pain and itch management: Analgesics, antihistamines, and neuropathic agents improve comfort but do not affect inflammatory or fibrotic drivers.
Surgical interventions: Contracture release, pseudosyndactyly separation, and oesophageal dilations temporarily address fibrotic sequelae but fibrosis often recurs quickly.
Nutritional support: High-calorie, high-protein diets and supplements counteract catabolism from systemic inflammation but cannot reverse it.
Experimental/Off-label Medications: several clinical studies and case reports have been published (more below)
Limitations
No disease modification: Current care does not interrupt persistent IL-1β, IL-6, TNF, TGF-β or NF-κB signalling — the suspected core pathways sustaining inflammation and fibrosis in RDEB.
Recurrence is inevitable: Surgical corrections for fibrotic deformities often need repeating, as the underlying drivers remain active.
Systemic inflammation unchecked: Supportive measures may reduce infection-related flares but do little to address the baseline cytokine storm that drives anaemia, growth delay, and metabolic decline.
Late-stage cancer risk remains: Even optimal standard care does not significantly reduce the high lifetime risk of SCC emerging from chronically inflamed, fibrotic skin.
The reality is that today’s standard-of-care manages symptoms, not mechanisms. Until treatments can directly modulate inflammatory signalling or fibrotic activation, RDEB patients remain locked in the injury–inflammation–fibrosis cycle — with predictable, devastating consequences.
6. Pipeline & emerging science
While no approved treatments yet interrupt the inflammatory–fibrotic cycle in RDEB, several approaches in the preclinical and clinical stages are aiming to do exactly that. The common theme: modulating the wound microenvironment to calm chronic inflammation, blunt fibroblast over-activation, and improve tissue repair.
Mesenchymal Stromal Cells (MSCs)
Two clinical-stage programmes — Rheacell (allo-MSCs) and Mission EB (allo-MSCs) — are exploring systemic delivery of MSCs in RDEB. Beyond their regenerative potential, MSCs are potent immunomodulators: they secrete anti-inflammatory cytokines (e.g. IL-10, TGF-β3), inhibit T-cell proliferation, and skew macrophages toward pro-resolving phenotypes. Early studies in RDEB have indicated improvements in wound healing speed, reduced pain and itch, and better skin integrity — effects likely arising more from immune modulation than from durable engraftment of new fibroblasts or keratinocytes. However, MSC therapy is transient, requiring repeat dosing, and overall response, the magnitude/duration of anti-inflammatory benefit in severe disease is still being defined.
Below you can see Sharmila Collins (Cure EB), Dr Anna Martinez (GOSH) & the Freir family discuss the results of the Mission EB trial.
Separately, in preclinical studies on RDEB model mice, systemically administered human cord blood-derived unrestricted somatic stem cells, which are considered precursors of MSCs, were effective in providing symptomatic relief, including the reduction of fibrotic mitten deformities. One mechanism involved was the modulation of IL-1-mediated inflammation via secretion of leukemia inhibitory factor (LIF).
Repurposing — Pathway-Targeted Approaches
Given the clear cytokine and signalling profile in RDEB, several well-characterised pathways stand out for therapeutic repurposing:
IL-1 axis: IL-1β is an upstream driver of NF-κB activation and inflammatory fibroblast phenotype. Blockade with agents like anakinra (IL-1 receptor antagonist) or canakinumab (anti–IL-1β mAb) has strong mechanistic rationale and preclinical validation in related skin inflammation models. However, low half-life of anakinra requiring frequent dosing is a limitation.
IL-6 axis: Targeting IL-6 with tocilizumab (anti–IL-6R) or similar biologics could reduce systemic inflammation, anaemia of chronic disease, and Th17 skewing.
TNF: Anti-TNF agents are proven in other chronic inflammatory dermatoses, though infection risk is a consideration in RDEB.
TGF-β: Inhibitors of TGF-β activation (e.g. anti–thrombospondin-1 approaches, fresolimumab) have potential to limit fibrosis while sparing wound closure if dosed carefully.
Losartan — Anti-Fibrotic with Anti-Inflammatory Side Benefits
Losartan, an angiotensin II receptor blocker that via multiple mechanisms reduces inflammation, limits fibrosis and inflammatory signaling in RDEB mouse models. Clinically, a small adult case series showed better severity scores, quality of life, and reduction of dermal collagen, while the REFLECT Phase 1/2 trial in 29 children confirmed safety and suggested benefits in EBDASI, quality of life, pain, itch, growth, skin elasticity, and reduction of TNF. However, disease stage and severity appeared to majorly influence the response.
JAK Inhibitors (JAKi’s)
Given the activation of multiple JAK–STAT pathways downstream of IL-6, IFN-γ, and other cytokines in RDEB, JAK inhibitors (e.g. ruxolitinib, baricitinib) offer a way to intercept broad inflammatory signalling. Topical and systemic JAKi’s are being explored and approved for other chronic skin diseases and could, in theory, dampen both immune-driven inflammation and fibroblast activation. Infection risk and wound healing impact would require close monitoring in RDEB trials.
Cannabinoids (CBD / THC)
Cannabinoid signaling involves CB1 receptors, mainly in the CNS, and CB2 receptors, largely in the immune system; THC primarily activates CB1 for analgesia, while CBD interacts more with CB2 to reduce inflammation. Both receptors are also present in skin cells and sensory fibers, and cannabinoids may aid wound healing by enhancing keratinocyte migration, increasing PDGF-A/B, and modulating cytokines (↓IL-1β, IL-6, TNF, TGF-β1; ↑IFN-γ). In EB, cannabinoid-based medicines have been reported to relieve pain and itch, with a survey of 71 patients suggesting benefits from topical and oral use, though evidence is limited and product variability complicates potential treatment.
Other Approaches
Matrix therapeutics: Agents that alter the extracellular matrix or are based on it (e.g. decorin mimetics) could both sequester TGF-β and normalise fibroblast behaviour.
Microbiome-directed therapy: Targeting S. aureus dominance with bacteriophage therapy or topical probiotics could reduce inflammasome activation and downstream IL-1β signalling.
Repurposed small molecules: evaluation of small molecules with established efficacy in other indications relevant to chronic wounds and dermatological fibrosis.
Exosome-based delivery: MSC-derived or engineered exosomes can carry anti-inflammatory cytokines or RNA cargo to wounds, reprogramming fibroblasts and immune cells.
Gene modulation of inflammatory mediators: RNAi or antisense approaches to knock down key cytokines or activators in skin fibroblasts or keratinocytes are early but conceptually attractive.
Angiotensin-(1–7): An anti-inflammatory peptide that has shown efficacy in RDEB mouse models, reducing fibrosis and inflammatory gene expression.
IL-17 inhibition: IL-17 is strongly upregulated in RDEB skin, fueling fibroblast activation and MMP induction. Anti–IL-17 biologics (e.g. secukinumab) may blunt this loop.
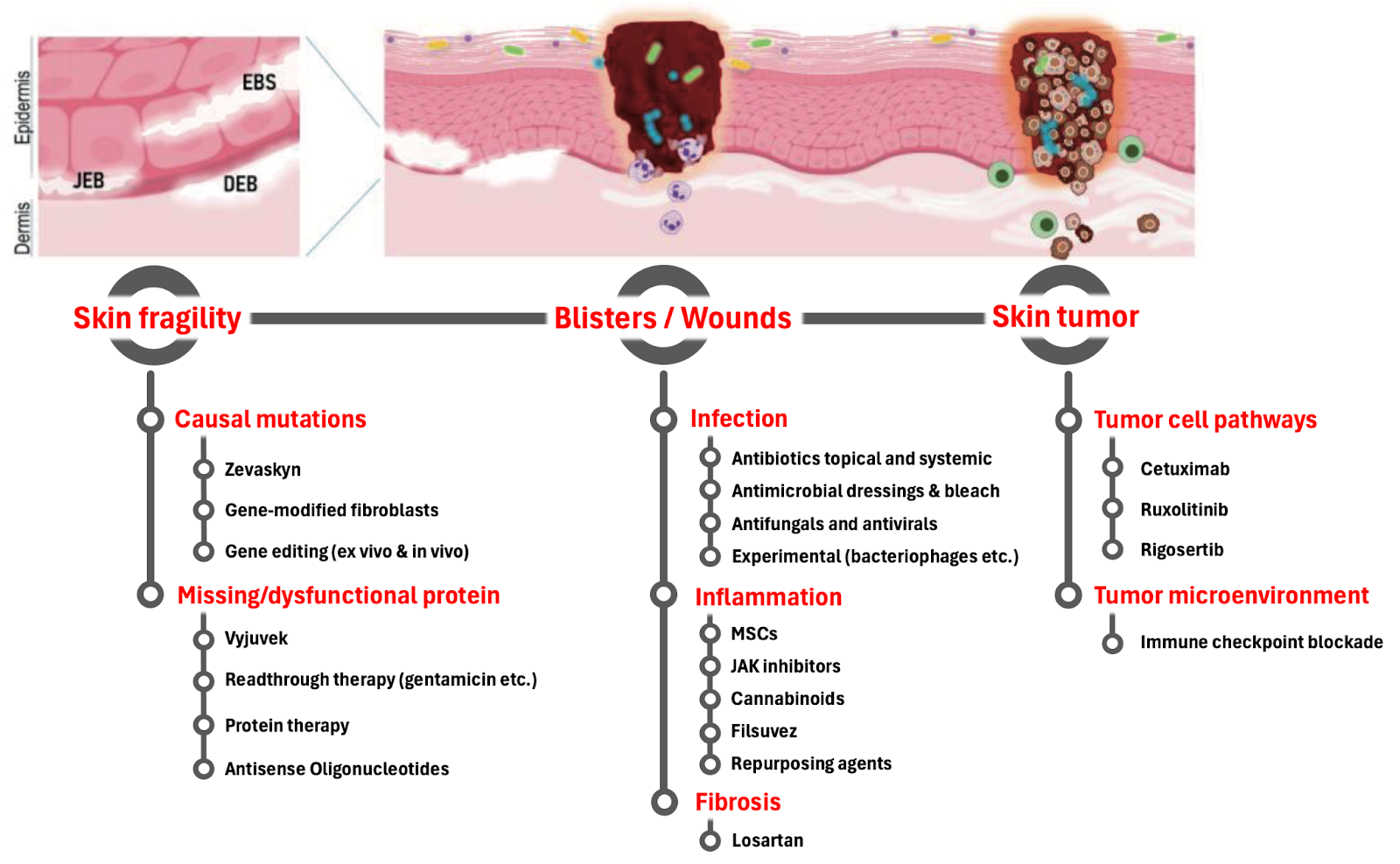
Fig. 5. The different therapeutic strategies targeting important aspects of the disease including inflammation and fibrosis. Adopted with changes from Hofbauer et. al. Chapter in Rare Diseases - Diagnostic and Therapeutic Odyssey, 2020
Taken together, these strategies represent a shift from purely supportive care toward mechanism-driven intervention — treating the underlying inflammatory and fibrotic biology of RDEB, not just its downstream complications. The challenge will be balancing efficacy with safety in fragile skin, and determining how best to combine anti-inflammatory and anti-fibrotic approaches for durable benefit.
7. Opportunities & challenges
Opportunities
Established disease mechanisms: Research over several decades has identified chronic inflammatory (IL-1β, IL-6, TNF, NF-κB) and fibrosis (TGF-β1, αSMA⁺ fibroblasts) pathways as central drivers of RDEB
Repurposing potential: Multiple approved drugs (e.g. losartan, JAKi’s, anti-IL-1, anti-IL-6) have strong mechanistic rationale and known safety profiles.
Measurable biomarkers: Cytokine levels, wound size, fibrosis markers, and functional scores can provide objective readouts in trials.
High unmet need: No disease-modifying options exist, creating strong patient, clinician, and payer interest in effective solutions.
Orphan disease incentives: RDEB qualifies for Orphan Drug status. Accelerated pathways like Fast Track or Breakthrough Therapy can shorten development timelines. Premium pricing for ultra-rare diseases supports reimbursement at six to seven figures annually, allowing returns even with small patient numbers.
Synergy with gene therapies: Most emerging RDEB therapies are gene or cell based, aiming to restore C7. Anti-fibrotic or anti-inflammatory drugs could boost their effect, e.g., systemic anti-TGF-β to prevent graft scarring.
Broadening horizons: RDEB shares fibrosis pathways with other indications e.g. scleroderma and keloids. A therapy proven in RDEB could expand to larger fibrotic disease markets.
Challenges
Fragile skin & safety risk: Targeting wound-healing pathways may compromise normal repair and immune surveillance. Systemic immunosuppression can worsen infection risk in open wounds.
Heterogeneous disease severity: Variability in RDEB phenotypes makes patient selection and endpoint design complex.
Chronic nature of pathology: Inflammation and fibrosis are self-perpetuating, so interventions may need long-term or combination therapy.
8. Market size
For high-unmet-need rare diseases with chronic dosing, annual pricing can range $150k–$500k (examples: IL-1β mAbs, rare dermatology biologics, gene therapy maintenance meds).
Pan-EB opportunity: An anti-inflammatory that works in RDEB may also benefit severe JEB or EBS with chronic wounds → adds perhaps 20–30% to the targetable population.
Label creep / repurposing leverage: Many of the candidate pathways (IL-1, IL-6, JAK/STAT, TGF-β) are implicated in other fibrotic and inflammatory diseases (systemic sclerosis, chronic wounds, scleroderma, etc.). A successful RDEB proof-of-concept could derisk expansion into much larger indications.
Competition in EB is centered on gene and cell therapies (Krystal, Abeona, Castle Creek). No marketed anti-fibrotic or anti-inflammatory therapies exist—these would be complementary, not competitive, and could establish themselves as standard-of-care. Such drugs could also expand the DEB market and open opportunities in higher-prevalence indications like scleroderma and keloids.
9. Investor take-aways
High unmet need: No approved therapies directly targeting inflammation or fibrosis in RDEB; current care is purely supportive.
Defined biology: Well-mapped cytokine and fibrotic pathways (IL-1β, IL-6, TNF, NF-κB, TGF-β1) offer clear mechanistic targets.
Repurposing advantage: Multiple approved drugs (losartan, JAKi’s, IL-1/IL-6 blockers) have strong rationale and known safety profiles.
Market leverage: RDEB offers rare disease entry with further expansion to pan-EB market and broader anti-fibrotic or anti-inflammatory indications, adjunct use with gene/cell therapies, and adjacent fibrosis indications.
Value inflection potential: Biomarker-rich, small patient trials could deliver rapid proof-of-concept and platform expansion opportunities.
Next up: Inflammation-Driven Itch in DEB